Autoinflammatorische Syndrome
Autoinflammatorische Syndrome (AIS) umfassen relativ seltene monogenetische Erkrankungen mit periodisch auftretenden Symptomen.
Die Serumwerte der Entzündungsproteine (C-reaktives Protein (CRP) und Serum-Amyloid-A (SAA)) können in einem Schub stark ansteigen und zwischen den Schüben normal sein. Bei Verdacht auf ein periodisches Syndrom sollten die CRP-Werte im Schub dokumentiert werden. Bei einem klinischen Verdacht auf ein AIS und erhöhten CRP-Werten im Schub kann die Diagnose durch einen Gentest bestätigt werden. Je nach Entität können Symptome ab der Geburt oder erst im Erwachsenenalter auftreten (Tab. 1). Häufig besteht eine typische Familienanamnese, es wurden aber auch Spontanmutationen beschrieben. Bei einem bestehenden klinischen Verdacht, sollte versucht werden, die Diagnose durch einen Gentest zu sichern. Eine klinische Differenzierung der periodischen Syndrome kann durch das Alter bei Erstmanifestation, die Dauer der Schübe und die Kombination der einzelnen Symptome im Schub erfolgen (Tab. 1). Bei vielen Patienten bestehen auch zwischen den Schüben leicht erhöhte CRP-Werte.
Jede chronisch-entzündliche Erkrankung kann zu einer Amyloidose führen. Bei den verschiedenen AIS erkranken ca. 30% aller Patienten im Laufe Ihres Lebens an einer Amyloidose vom Typ AA.
Familiäres Mittelmeerfieber
Das familiäre Mittelmeerfieber (FMF) ist das weltweit häufigste periodische Syndrom [3, 9, 28]. Im Schub bestehen Bauchschmerzen, thorakale Schmerzen und deutlich erhöhte CRP-Werte. Fieber kann bei Beginn eines Schubes vorhanden sein, tritt aber nicht bei allen Patienten und nicht in jedem Schub auf. Oft werden in Unkenntnis der Diagnose eine Appendektomie oder eine Laparoskopie mit unspezifischen oder unauffälligen Befunden durchgeführt. Weitere typische Symptome sind Gelenkschmerzen und Hauträtungen. Die meisten Patienten stammen aus der Türkei, Armenien, Israel, Libanon, Marokko und anderen Ländern des Mittelmeerraumes. Bei Deutschen Patienten ist die Erkrankung sehr selten. Die klinische Diagnose kann durch Nachweis von Mutationen im MEFV-Gen bestätigt werden. Patienten mit einer homozygoten M694V-Mutation sind jünger bei Krankheitsbeginn und haben häufiger Gelenkbeschwerden.
Ob Patienten mit dieser Mutation ein höheres Risiko für die Entwicklung einer AA-Amyloidose haben wird von einigen Autoren behauptet [3], von anderen Autoren jedoch nicht bestätigt [18]. Die Standardtherapie des familiären Mittelmeerfiebers ist eine Dauertherapie mit Colchicum [3, 9, 28]. Unter Colchicum treten deutlich weniger und mildere Schübe auf. Das gute Ansprechen auf eine Therapie mit Colchicum kann die Diagnose in unklaren Fällen unterstützen und ist eines von mehreren etablierten Diagnosekriterien [18]. Auch Patienten mit einem partiellen Ansprechen auf eine Colchicum-Therapie sollten regelmäßig Colchicum zur Prävention einer AA-Amyloidose einnehmen. Das familiäre Mittelmeerfieber (FMF) ist das weltweit häufigste periodische Syndrom [3, 9, 28]. Im Schub bestehen Bauchschmerzen, thorakale Schmerzen und deutlich erhöhte CRP-Werte. Fieber kann bei Beginn eines Schubes vorhanden sein, tritt aber nicht bei allen Patienten und nicht in jedem Schub auf. Oft werden in Unkenntnis der Diagnose eine Appendektomie oder eine Laparoskopie mit unspezifischen oder unauffälligen Befunden durchgeführt. Weitere typische Symptome sind Gelenkschmerzen und Hauträtungen. Die meisten Patienten stammen aus der Türkei, Armenien, Israel, Libanon, Marokko und anderen Ländern des Mittelmeerraumes. Bei Deutschen Patienten ist die Erkrankung sehr selten. Die klinische Diagnose kann durch Nachweis von Mutationen im MEFV-Gen bestätigt werden. Patienten mit einer homozygoten M694V-Mutation sind jünger bei Krankheitsbeginn und haben häufiger Gelenkbeschwerden. Ob Patienten mit dieser Mutation ein höheres Risiko für die Entwicklung einer AA-Amyloidose haben wird von einigen Autoren behauptet [3], von anderen Autoren jedoch nicht bestätigt [18]. Die Standardtherapie des familiären Mittelmeerfiebers ist eine Dauertherapie mit Colchicum [3, 9, 28]. Unter Colchicum treten deutlich weniger und mildere Schübe auf. Das gute Ansprechen auf eine Therapie mit Colchicum kann die Diagnose in unklaren Fällen unterstützen und ist eines von mehreren etablierten Diagnosekriterien [18]. Auch Patienten mit einem partiellen Ansprechen auf eine Colchicum-Therapie sollten regelmäßig Colchicum zur Prävention einer AA-Amyloidose einnehmen.

Abb. 1: FMF: Peritonitis-Schübe über 2-5 Tage.
Cryopyrin-assoziierte periodische Syndrome
Die Cryopyrin assoziierten periodischen Syndrome (CAPS) umfassen das familiäre Kälte-assoziierte autoinflammatorische Syndrom (Kälteurtikaria) [27], das Muckle-Wells-Syndrom [21] und das chronische infantile neuro-cutane Arthritis-Syndrom (CINCA) [22]. Diese Syndrome sind mit Mutationen im CIAS1-NLRP3-Gen assoziiert [1, 8] und stellen ein Krankheitsspektum mit milden (Kälteurtikaria), moderaten (Muckle-Wells) und schweren Verläufen (CINCA) dar. Die klinischen Symptome umfassen Hautrötungen, eine Nesselsucht der Haut, Schwerhörigkeit, Bindehautentzündungen der Augen und häufige Kopfschmerzen. Das CRP ist im Schub immer deutlich erhöht. Das CIAS1/NALP3-Gen codiert das Protein Cryopyrin, Mutationen führen zu einer exzessiven Überproduktion von IL-1?. Bei CINCA-Patienten können zusätzlich Fehlbildungen des Knochen- und Knorpelwachstums mit massiven Exostosen und einer Patellahypertrophie auftreten. Die Therapie erfolgt mit Biologika die gegen das Interleukin (IL)-1? gerichtet sind (Tab. 2).
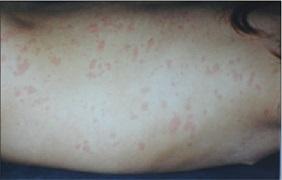
Abb. 2: Urtikarielles Exanthem bei CAPS
TRAPS
Das TNF-Rezeptor assoziierte periodische Syndrom (TRAPS, hibernisches Fieber [30]) ist mit Mutationen im TNFRSF1A-Gen assoziiert [19]. Die klinischen Symptome sind ähnlich dem familiären Mittelmeerfieber (Tab. 1), eine Colchicum-Therapie ist unwirksam. Im Schub können starke Muskelschmerzen auftreten. Im Schub bestehen deutlich erhöhte CRP-Werte. Die CRP-Werte sind auch zwischen den symptomatischen Episoden bei einigen Patienten deutlich erhöht. Eine Assoziation von TRAPS und demyelinisierenden neurologischen Erkrankungen wurde beschrieben [14]. Bei mittelstarken Verläufen kann ein Therapieversuch mit Steroiden, Methotrexat oder Azathioprin unternommen werden. Bei Unwirksamkeit haben einige Autoren eine gute Wirksamkeit von Antikörpern gegen TNF? beschrieben [7, 13], bei manchen Patienten wurden aber auch Schübe durch anti-TNF?-Medikamente ausgelöst [12]. Kürzlich wurde gezeigt, dass anti-IL1?-Medikamente bei einigen Patienten mit TRAPS besser wirksam sind als andere Biologika.
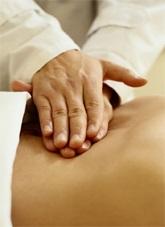
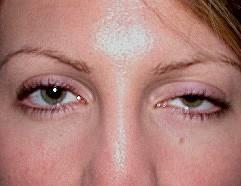
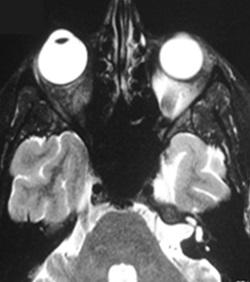
Abb. 3: TRAPS: Bauchschmerzen, hängendes Augenlid und Muskelschmerzen mit Muskelödem im MRT.
HIDS
Das Hyper-IgD-Syndrom („Holländisches Fieber“) und die Mevalonatkinase-Defizienz sind beide auf Mutationen des Mevalonatkinase-Genes zurückzuführen [10]. Die Fieberschübe sind von cervikalen Lymphnotenschwellungen, Exanthemen, Bauchschmerzen, Diarrhoe, Gelenkschmerzen und oralen Ulcera begleitet. Im Serum finden sich erhöhte Konzentrationen von Immunglobulinen vom Typ IgD, jedoch nicht bei allen Patienten.
Die Mevalonatkinase-Defizienz ist eine Stoffwechselerkrankung, bei der die Mevalonsäure-ausscheidung im Urin erhöht und das Cholesterin im Serum aufgrund der Synthesestörung deutlich vermindert ist. Auch hierbei sind die IgD-Werte deutlich erhöht. Die Diagnose kann durch Nachweis einer Mutation im MVK-Gen gesichert werden. Die Therapie erfolgt mit leichten Fällen mit nicht-steroidalen Antirheumatica, in schweren Fällen mit Antikörpern gegen TNF? oder IL-1? (Tab. 2), Colchicum ist unwirksam.
Seltene periodische Syndrome
Das Pyodermie-Akne-Pustulose-Arthritis (PAPA)-Syndrom [17] ist mit einer Mutation des CD2BP1-Genes assoziiert. In den wenigen bekannten Fällen wurden Antikörper gegen TNF? erfolgreich eingesetzt.
Das Blau-Syndrom (BS) ist eine familiäre Sarkoidose mit Beginn in der frühen Kindheit [5]. Die juvenile Sarkoidose ist eine sporadisch auftretende Erkrankung [25]. Beide Krankheiten sind mit Mutationen im CARD-15-Gen assoziiert [20]. Zu den klinischen Symptomen gehören Fieberschübe, Exantheme, kutane Granulome, eine granulomatöse Synovitis und eine Uveitis. Im Gegensatz zur Sarkoidose der Erwachsenen besteht keine Lungenbeteiligung. Im Langzeitverlauf stehen Kontrakturen der Finger und der Zehen (Camptodaktylie) und ein schweres Sekundärglaukom im Vordergrund. Wirksame Medikamente sind Methotrexat, Leflunomid und Biologika [2].
Das Periodische Fieber Aphthöse Stomatitis Pharyngitis und Adenitis (PFAPA) Syndrom ist eine relativ häufige Erkrankung die in der Regel in der Kindheit auftritt und durch sehr regelmäßig auftretendes Fieber, Halsschmerzen und Aphten gekennzeichnet ist. Im Schub sind die CRP-Werte deutlich erhöht. Im Gegensatz zu den anderen AIS ist beim PFAPA (noch) keine Genetische Mutation bekannt. Durch eine Tonsillektomie können ca. 30% der Patienten in eine Remission gebracht werden. Im Schub kann durch einen Steroidstoß eine Reduktion der Intensität und der Schubdauer erreicht werden. Bei häufigen Schüben könnte eine Therapie mit Kineret ebenfalls wirksam sein. In den meisten Fällen klingen die Schübe nach der Pubertät ab. Persistierende Schübe im Erwachsenenalter wurden beschrieben, sind aber selten.
| Familiäres Mittelmeer-Fieber | Hyper-IgD-Syndrom | TNF Rezeptor assoziiertes periodisches Syndrom | Muckle-Wells-Syndrom | Familiäre Kälteurticaria | Chronisch-infantiles neuro-kutanes Arthritis-Syndrom | Pyogene Arthritis Pyodermie Akne-Syndrom | Blau-Syndrom juvenile Sarkoidose | |
|---|---|---|---|---|---|---|---|---|
| Alter bei Beginn | < 10 Jahre | < 1 Jahr | < 20 Jahre | Kindheit | Kindheit | < 1 Monat | <10 Jahre | < 1 Jahr |
| Fieberdauer | 1-3 Tage | 3-7 Tage | >7 Tage | Tage-Wochen | Tage-Wochen | variabel | variabel | variabel |
| freies Intervall | Wochen-Monate | 4-8 Wochen | Monate | variabel | variabel | variabel | variabel | variabel |
| Klinik | Polyserositis | cervikale Lymphknoten Erbrechen Diarrhoe | periorbitale Ödeme Konjunktivitis | Hörstürze | Kälte-Urtikaria | Meningitis | pyogene Arthritis Pyoderma Akne | Granulome |
| Peritonitis | Taubheit | Urtikaria Arthritis | Iridozyklitis | |||||
| Exostosen | Glaukom | |||||||
| Gelenke | Monarthritis | Arthritis | Myalgien | keine | keine | Arthritis | Arthritis | Polyarthritis |
| Kontrakturen | ||||||||
| Haut | Erysipel | Erythem | Erythem | Urtikaria | Urtikaria | Urtikaria | Pyodermie | Erythem |
| Amyloidose | ja | nein | ja | ja | nein | ja | nein | nein |
| Gen | MEFV | MVK | TNFRSF1A | CIAS1 | CIAS1 | CIAS1 | CD2BP1 | CARD-15 |
| Protein | Pyrin | MVK | TNFRezeptor | Cryopyrin | Cryopyrin | Cryopyrin | PSTPIP | CARD-15 |
| Therapie | Colchicin | Anakinra | Steroide | Anakinra | Anakinra | Anakinra | Steroide DMARD | Steroide DMARD |
| Anti-TNF? | Canakinumab | Rilonacept | Canakinumab | Anti-TNF? | Anti-TNF? | |||
| Anakinra | Canakinumab | |||||||
| Manifestation | benigne Peritonitis | Hyper gD dutch fever | hibernisches Fieber | Schwer-hörigkeit | Kälte-Urticaria | CINCA | PAPA | juvenile Sarkoidose |
| Jahr der Erst-beschreibung | 1945 | 1984 | 1982 | 1962 | 1969 | 1987 | 1997 | 1984 |
| Autor | Siegal | Van der Meer | Wiliamson | Muckle | Tindall | Prieur | Lindor | Blau |
| Zielstruktur | Medikamententyp | |
|---|---|---|
| Etanercept | TNF-? | TNF-Rezeptor-IgG-Konstrukt |
| Infliximab | TNF-? | chimärer monoklonaler Antikörper |
| Adalimumab | TNF-? | humaner monoklonaler Antikörper |
| Golimumab | TNF-? | humaner monoklonaler Antikörper |
| Certolizumab | TNF-? | pegyliertes Fab2-Antikörperfragment |
| Anakinra | IL-1b | rekombinant humaner IL-1-Rezeptorantagonist |
| Rilonacept | IL-1b | IL-1-Rezeptor-IgG-Konstrukt |
| Canakinumab | IL-1b | humaner monoklonaler Antikörper |
| Tocilizumab | IL-6R | humanisierter monoklonaler Antikörper |
| Rituximab | CD20 | chimärer monoklonaler Antikörper |
| Abatacept | B7 | CTLA4-IgG-Konstrukt, blockiert Kostimulation |
Literatur
- Aksentijevich I, Nowak M, Mallah M, et al. (2002). De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum. 46:3340-3348.
- Aróstegui JI, Arnal C, Merino R, et al. (2007). NOD2 gene-associated pediatric granulomatous arthritis: clinical diversity, novel and recurrent mutations, and evidence of clinical improvement with interleukin-1 blockade in a Spanish cohort. Arthritis Rheum. 56:3805-3813.
- Ben-Chetrit E, Levy M (1998). Familial Mediterranean fever. Lancet 351:659-664.
- Bergesio F, Ciciani AM, Manganaro M, et a., (2008). Renal involvement in systemic amyloidosis: an Italian collaborative study on survival and renal outcome. Nephrol Dial Transplant. 23:941-951.
- Blau EB (1985). Familial granulomatous arthritis, iritis, and rash. J Pediatr. 107:689-693.
- Dember LM, Hawkins PN, Hazenberg BP, et al. (2007). Eprodisate for the treatment of renal disease in AA amyloidosis. N Engl J Med. 356:2349-2360.
- Drewe E, McDermott EM, Powell PT, Isaacs JD, Powell RJ (2003). Prospective study of anti-tumour necrosis factor receptor superfamily 1B fusion protein, and case study of anti-tumour necrosis factor receptor superfamily 1A fusion protein, in tumour necrosis factor receptor associated periodic syndrome (TRAPS): clinical and laboratory findings in a series of seven patients. Rheumatology. 42:235-239.
- Feldmann J, Prieur AM, Quartier P, Berquin P, Certain S, Cortis E, Teillac-Hamel D, Fischer A, de Saint Basile G (2002). Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am J Hum Genet. 71:198-203.
- Fonnesu C, Cerquaglia C, Giovinale M, et al. (2008). Familial Mediterranean Fever: A review for clinical management. Joint Bone Spine. [Epub ahead of print]
- Haas D, Hoffmann GF (2006). Mevalonate kinase deficiencies: from mevalonic aciduria to hyperimmunoglobulinemia D syndrome. Orphanet J Rare Dis. 1:13. Review.
- Hazenberg BP, Bijzet J, Limburg PC et al. (2007). Diagnostic performance of amyloid A protein quantification in fat tissue of patients with clinical AA amyloidosis. Amyloid. 14:133-140.
- Jacobelli S, André M, Alexandra JF, Dodé C, Papo T (2007). Failure of anti-TNF therapy in TNF Receptor 1-Associated Periodic Syndrome (TRAPS). Rheumatology. 46:1211-1212.
- Kallinich T, Haffner D, Rudolph B, et al. (2006). „Periodic fever“ without fever: two cases of non-febrile TRAPS with mutations in the TNFRSF1A gene presenting with episodes of inflammation or monosymptomatic amyloidosis. Ann Rheum Dis. 65:958-960.
- Kümpfel T, Hoffmann LA, Rübsamen H, Pöllmann W, Feneberg W, Hohlfeld R, Lohse P (2007). Late-onset tumor necrosis factor receptor-associated periodic syndrome in multiple sclerosis patients carrying the TNFRSF1A R92Q mutation. Arthritis Rheum. 56:2774-2783.
- Koivuniemi R, Paimela L, Suomalainen R, Tornroth T, Leirisalo-Repo M. (2008). Amyloidosis is frequently undetected in patients with rheumatoid arthritis. Amyloid. 15:262-268.
- Lachmann HJ, Goodman HJ, Gilbertson JA, Gallimore JR, Sabin CA, Gillmore JD, Hawkins PN. (2007). Natural history and outcome in systemic AA amyloidosis. N Engl J Med. 356:2361-2371.
- Lindor NM, Arsenault TM, Solomon H, Seidman CE, McEvoy MT (1997). A new autosomal dominant disorder of pyogenic sterile arthritis, pyoderma gangrenosum, and acne: PAPA syndrome. Mayo Clin Proc. 72:611-615.
- Livneh A, Langevitz P, Zemer D, et al. (1997). Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum. 40:1879-1885.
- McDermott MF, Aksentijevich I, Galon J, et al. (1999). Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell. 97:133-144.
- Miceli-Richard C, Lesage S, Rybojad M, Prieur AM, Manouvrier-Hanu S, Häfner R, Chamaillard M, Zouali H, Thomas G, Hugot JP (2001). CARD15 mutations in Blau syndrome. Nat Genet. 29:19-20.
- Muckle TJ, Wells M (1962). Urticaria, deafness, and amyloidosis: a new heredo-familial syndrome. Q J Med. 31:235-248.
- Prieur AM, Griscelli C, Lampert F, Truckenbrodt H, Guggenheim MA, Lovell DJ, Pelkonnen P, Chevrant-Breton J, Ansell BM (1987). A chronic, infantile, neurological, cutaneous and articular (CINCA) syndrome. A specific entity analysed in 30 patients. Scand J Rheumatol Suppl. 66:57-68.
- Röcken C, Shakespeare A. (2002) Pathology, diagnosis and pathogenesis of AA amyloidosis. Virchows Arch. 440:111-122.
- Röcken C, Ernst J, Hund E, Michels H, Perz J, Saeger W, Sezer O, Spuler S, Willig F, Schmidt HH (2006). Interdisciplinary guidelines for diagnosis and therapy of extracerebral amyloidosis: issued by the German Society of Amyloid Diseases e. V. (www.amyloid.de). Med Klin. 101:825-829.
- Rosé CD, Doyle TM, McIlvain-Simpson G, Coffman JE, Rosenbaum JT, Davey MP, Martin TM (2005). Blau syndrome mutation of CARD15/NOD2 in sporadic early onset granulomatous arthritis. J Rheumatol. 32:373-375.
- Schönland SO (2006). Fortschritte in der Diagnostik und Therapie der Amyloidosen. Deutsches Ärzteblatt. 34-35:2237-2244.
- Tindall JP, Beeker SK, Rosse WF (1969). Familial cold urticaria. A generalized reaction involving leukocytosis. Arch Intern Med. 124:129-134.
- Tunca M, Akar S, Onen F et al. Turkish FMF Study Group (2005). Familial Mediterranean fever (FMF) in Turkey. Medicine 84:1-11.
- Vickery S, Webb MC, Price CP, John RI, Abbas NA, Lamb EJ (2008). Prognostic value of cardiac biomarkers for death in a non-dialysis chronic kidney disease population. Nephrol Dial Transplant. 23:3546-3553.
- Williamson LM, Hull D, Mehta R, Reeves WG, Robinson BH, Toghill PJ (1982). Familial Hibernian fever. Q J Med. 51:469-80.
Kontakt
Rheumazentrum-Heidelberg
Medizinische Klinik (Krehl-Klinik)
Prof. Dr. med. H. M. Lorenz
Im Neuenheimer Feld 410
69120 Heidelberg
Tel.: +49 6221 56-8030
E-Mail: info@rheumazentrum-heidelberg.de